The Human Pangenome Reference Consortium has introduced a significantly more diverse reference human genome sequence, the “pangenome,” composed of 94 distinct genome sequences and aiming to reach 700 by 2024, which helps in identifying larger genomic variants and enhancing genomic analysis accuracy.
More complete and sophisticated collection of genome sequences captures significantly more human diversity.
Researchers have released a new high-quality collection of reference human genome sequences that captures substantially more diversity from different human populations than what was previously available. The work was led by the international Human Pangenome Reference Consortium, a group funded by the National Human Genome Research Institute (NHGRI), part of the National Institutes of Health.
The new “pangenome” reference includes genome sequences of 47 people, with the researchers pursuing the goal of increasing that number to 350 by mid-2024. With each person carrying a paired set of chromosomes, the current reference actually includes 94 distinct genome sequences, with a goal of reaching 700 distinct genome sequences by the completion of the project.
The work, published in the journal Nature, is one of several papers published by consortium members.

A genome is the set of DNA instructions that helps each living creature develop and function. Genome sequences differ slightly among individuals. In the case of humans, any two peoples’ genomes are, on average, more than 99% identical. The small differences contribute to each person’s uniqueness and can provide insights about their health, helping to diagnose disease, predict outcomes and guide medical treatments.
“Everyone has a unique genome, so using a single reference genome sequence for every person can lead to inequities in genomic analyses. For example, predicting a genetic disease might not work as well for someone whose genome is more different from the reference genome.”
To understand these genomic differences, scientists create reference human genome sequences for use as a “standard” — a digital amalgamation of human genome sequences that can be used as a comparison to align, assemble and study other human genome sequences.
The original reference human genome sequence is nearly 20 years old and has been regularly updated as technology advances and researchers fix errors and discover more regions of the human genome. However, it is fundamentally limited in its representation of the diversity of the human species, as it consists of genomes from only about 20 people, and most of the reference sequence is from only one person.
“Everyone has a unique genome, so using a single reference genome sequence for every person can lead to inequities in genomic analyses,” said Adam Phillippy, Ph.D., senior investigator in the Computational and Statistical Genomics Branch within NHGRI’s Intramural Research Program and a co-author of the main study. “For example, predicting a genetic disease might not work as well for someone whose genome is more different from the reference genome.”
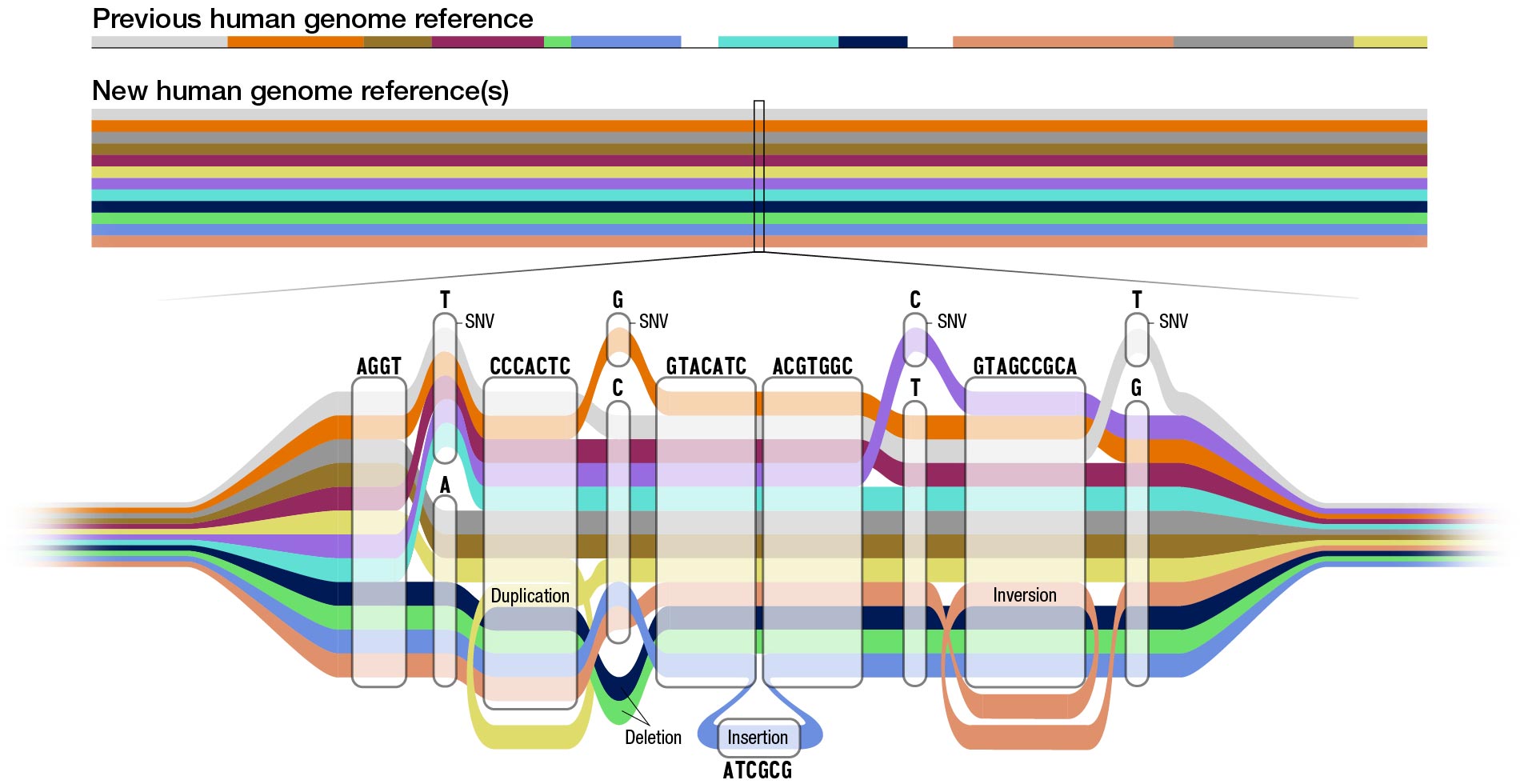
The current reference human genome sequence has gaps that reflect missing information, especially in areas that were repetitive and hard to read. Recent technological advances such as long-read DNA sequencing, which reads longer stretches of the DNA at a time, helped researchers fill in those gaps to create the first complete human genome sequence. This complete human genome sequence, released last year as part of the NIH-funded Telomere-to-Telomere (T2T) consortium, is incorporated into the current pangenome reference. In fact, many of the T2T researchers are also members of the Human Pangenome Reference Consortium.
Using advanced computational techniques to align the various genome sequences, the researchers constructed a new human pangenome reference with each assembly in the pangenome covering more than 99% of the expected sequence with more than 99% accuracy. It also builds upon the previous reference genome sequence, adding over 100 million new bases, or “letters” in DNA. While the previous reference genome sequence was single and linear, the new pangenome represents many different versions of the human genome sequence at the same time. This gives researchers a wider range of options for using the pangenome in analyzing other human genome sequences.
“By using the pangenome reference, we can more accurately identify larger genomic variants called structural variants,” said Mobin Asri, a Ph.D. student at the University of California Santa Cruz and co-first author of the paper. “We are able to find variants that were not identified using previous methods that depend on linear reference sequences.”
Structural variants can involve thousands of bases. Until now, researchers have been unable to identify the majority of structural variants that exist in each human genome using short-read sequencing due to the bias of using a single reference sequence.
“The human pangenome reference will enable us to represent tens of thousands of novel genomic variants in regions of the genome that were previously inaccessible,” said Wen-Wei Liao, a Ph.D. student at Washington University in St. Louis, research affiliate at Yale University and co-first author of the paper. “With a pangenome reference, we can accelerate clinical research by improving our understanding of the link between genes and disease traits.”
The total cost of supporting the work of the Human Pangenome Reference Consortium is projected to be about $40 million over five years, which includes efforts to create the human pangenome reference, improve DNA sequencing technology, operate a coordinating center, conduct outreach and create resources for the research community to use the pangenome reference.
Many of the individuals whose genomes were sequenced for constructing the new human pangenome reference were originally recruited as part of the 1,000 Genomes Project, a collaborative and international effort funded in part by NIH that aimed to improve the catalog of genomic variants in diverse populations. Because the human pangenome reference is a work in progress, researchers from the international Human Pangenome Reference Consortium continue to add more genome sequences to increasingly improve the quality of the pangenome reference.
“Basic researchers and clinicians who use genomics need access to a reference sequence that reflects the remarkable diversity of the human population. This will help make the reference useful for all people, thereby helping to reduce the chances of propagating health disparities,” said Eric Green, M.D., Ph.D., NHGRI director. “Creating and enhancing a human pangenome reference aligns with NHGRI’s goal of striving for global diversity in all aspects of genomics research, which is crucial to advance genomic knowledge and implement genomic medicine in an equitable way.”
In line with this effort, the Human Pangenome Reference Consortium includes an embedded ethics group that is working to anticipate challenging issues and help guide informed consent, prioritize the study of different samples, explore possible regulatory issues pertaining to clinical adoption, and work with international and Indigenous communities to incorporate their genome sequences in these broader efforts.
“A draft human pangenome reference” by Wen-Wei Liao, Mobin Asri, Jana Ebler, Daniel Doerr, Marina Haukness, Glenn Hickey, Shuangjia Lu, Julian K. Lucas, Jean Monlong, Haley J. Abel, Silvia Buonaiuto, Xian H. Chang, Haoyu Cheng, Justin Chu, Vincenza Colonna, Jordan M. Eizenga, Xiaowen Feng, Christian Fischer, Robert S. Fulton, Shilpa Garg, Cristian Groza, Andrea Guarracino, William T. Harvey, Simon Heumos, Kerstin Howe, Miten Jain, Tsung-Yu Lu, Charles Markello, Fergal J. Martin, Matthew W. Mitchell, Katherine M. Munson, Moses Njagi Mwaniki, Adam M. Novak, Hugh E. Olsen, Trevor Pesout, David Porubsky, Pjotr Prins, Jonas A. Sibbesen, Jouni Sirén, Chad Tomlinson, Flavia Villani, Mitchell R. Vollger, Lucinda L. Antonacci-Fulton, Gunjan Baid, Carl A. Baker, Anastasiya Belyaeva, Konstantinos Billis, Andrew Carroll, Pi-Chuan Chang, Sarah Cody, Daniel E. Cook, Robert M. Cook-Deegan, Omar E. Cornejo, Mark Diekhans, Peter Ebert, Susan Fairley, Olivier Fedrigo, Adam L. Felsenfeld, Giulio Formenti, Adam Frankish, Yan Gao, Nanibaa’ A. Garrison, Carlos Garcia Giron, Richard E. Green, Leanne Haggerty, Kendra Hoekzema, Thibaut Hourlier, Hanlee P. Ji, Eimear E. Kenny, Barbara A. Koenig, Alexey Kolesnikov, Jan O. Korbel, Jennifer Kordosky, Sergey Koren, HoJoon Lee, Alexandra P. Lewis, Hugo Magalhães, Santiago Marco-Sola, Pierre Marijon, Ann McCartney, Jennifer McDaniel, Jacquelyn Mountcastle, Maria Nattestad, Sergey Nurk, Nathan D. Olson, Alice B. Popejoy, Daniela Puiu, Mikko Rautiainen, Allison A. Regier, Arang Rhie, Samuel Sacco, Ashley D. Sanders, Valerie A. Schneider, Baergen I. Schultz, Kishwar Shafin, Michael W. Smith, Heidi J. Sofia, Ahmad N. Abou Tayoun, Françoise Thibaud-Nissen, Francesca Floriana Tricomi, Justin Wagner, Brian Walenz, Jonathan M. D. Wood, Aleksey V. Zimin, Guillaume Bourque, Mark J. P. Chaisson, Paul Flicek, Adam M. Phillippy, Justin M. Zook, Evan E. Eichler, David Haussler, Ting Wang, Erich D. Jarvis, Karen H. Miga, Erik Garrison, Tobias Marschall, Ira M. Hall, Heng Li and Benedict Paten, 10 May 2023, Nature.
DOI: 10.1038/s41586-023-05896-x
“Increased mutation rate and gene conversion within human segmental duplications” by Mitchell R. Vollger, Philip C. Dishuck, William T. Harvey, William S. DeWitt, Xavi Guitart, Michael E. Goldberg, Allison N. Rozanski, Julian Lucas, Mobin Asri, Human Pangenome Reference Consortium, Katherine M. Munson, Alexandra P. Lewis, Kendra Hoekzema, Glennis A. Logsdon, David Porubsky, Benedict Paten, Kelley Harris, PingHsun Hsieh and Evan E. Eichler, 10 May 2023. Nature.
DOI: 10.1038/s41586-023-05895-y
“Recombination between heterologous human acrocentric chromosomes” by Andrea Guarracino, Silvia Buonaiuto, Leonardo Gomes de Lima, Tamara Potapova, Arang Rhie, Sergey Koren, Boris Rubinstein, Christian Fischer, Human Pangenome Reference Consortium, Jennifer L. Gerton, Adam M. Phillippy, Vincenza Colonna and Erik Garrison, 10 May 2023, Nature.
DOI: 10.1038/s41586-023-05976-y
“Pangenome graph construction from genome alignment with minigraph-cactus” by Glenn Hickey, Jean Monlong, Jana Ebler, Adam M. Novak, Jordan M. Eizenga, Yan Gao, Human Pangenome Reference Consortium, Tobias Marschall, Heng Li and Benedict Paten, 10 May 2023, Nature Biotechnology.
DOI: 10.1038/s41587-023-01793-w